Greenleaf Regulatory Landscape Series
The Food and Drug Administration (FDA or the Agency) user fee programs help to provide funding for the Agency to achieve its mission of protecting the public health and providing safe and effective medical products to patients in the United States.1 The user fee programs provide the FDA with financial support to meet specified performance goals and commitments related to medical product submissions. These specific performance goals and commitments are negotiated and agreed upon between the FDA and industry in the years leading up to the reauthorizations and must be sent to Congress for review and approval.
All medical product user fees are renegotiated every five years. As the Prescription Drug User Fee Amendments (PDUFA VI), the Biosimilar User Fee Amendments (BsUFA II), and the Medical Device User Fee Amendments (MDUFA IV) are also set to expire on September 30, 2022, it is expected that all three user fee program extensions will be part of the same legislative package.2,3 The next set of authorizations for these programs will cover Fiscal Years 2023 to 2027.
Below is a summary of the current status of the PDUFA, BsUFA, and MDUFA negotiations as well as a description of the key commitments being discussed between the FDA and industry.
PDUFA
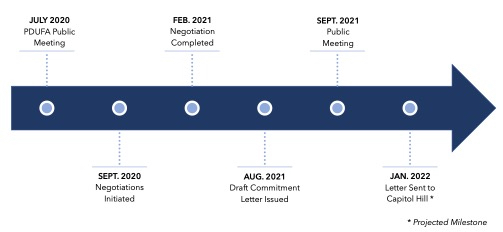
PDUFA VII will build upon the progress of past programs by providing the FDA with the resources and tools it needs to keep pace with advances in drug development. Considering recent scientific breakthroughs in cell and gene therapy and the bolus of investigational new drug (IND) applications for advanced biological therapies received by the Agency, PDUFA VII seeks to provide the Center for Biologics Evaluation and Research (CBER) with the funding and authority to hire additional staff to meet this demand. With negotiations being held virtually against the backdrop of the COVID-19 pandemic, PDUFA VII also aims to formalize some of the lessons learned from the pandemic, including guidance and workshops on the use of alternative tools to assess manufacturing facilities and additional resources to support the broader use of digital health tools (DHTs). The commitment letter,4 which was made public in late August 2021, includes the following commitments:
Strengthen Scientific Dialogue. Recognizing the need for further dialogue between the Agency and sponsors, the FDA will formalize the INTERACT meeting framework as well as establish a similar meeting type for the Center for Drug Evaluation and Research (CDER). Both industry and the FDA also discussed the possibility of sharing metrics related to all PDUFA meeting types and associated interactions.
Promote Innovation. To shorten review timelines for certain approved therapies and support efficacy endpoint development for rare diseases, PDUFA VII will establish the Split Real Time Application Review (STAR), modeled after CDER’s Real Time Oncology Review (RTOR).5
Support Advanced Biological Therapies. As noted, CBER will likely be provided dedicated resources to ensure the timely review of all applications for innovative biological therapies. Negotiations also focused on including the patient voice in gene therapy development programs as well as the potential for a workshop on how sponsors could leverage prior knowledge to accelerate gene therapy development. The FDA and industry also discussed a proposal on potential guidance dedicated to clarifying evidentiary standards for the RMAT program.6
Modernize Evidence Generation and Drug Development Tools. To advance the use of real-world evidence (RWE), both industry and the Agency discussed the establishment of a pilot program to develop new methods for using real-world data (RWD) in regulatory decision-making, including in the review of applications.7 Both industry and the FDA expressed an interest in continuing the Model Informed Drug Development (MIDD) paired meeting program. Issuance of guidance related to Complex Innovative Designs (CID) is also included in the commitment letter.8
Advance IT Infrastructure. PDUFA VII will also modernize data and information technology (IT) capacity and capabilities, including the adoption of cloud-based technologies as described in the FDA’s Technology Modernization Action Plan as well as technology convergence across the review centers more broadly.9 Both industry and the FDA also discussed potential programs and initiatives that could inform the evaluation of DHT-generated data.10
Next Steps
The initial draft of the PDUFA commitment letter has been published and is currently open to comment.11 The letter will also be reviewed and discussed at a public meeting on September 28, 2021.
PDUFA VII Negotiation Process
BsUFA
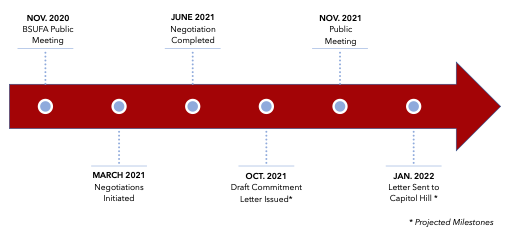
With negotiations recently completed on the heels of the PDUFA VII negotiations, BSUFA III will likely include several commitments that were agreed to in PDUFA VII, such as the development of guidance on alternative tools to assess manufacturing facilities, the dedication of resources to modernize the FDA’s data and information technology capabilities, and resources to enhance hiring and retention. Additional focus areas from the BSUFA III negotiations are outlined below:
Greater Collaboration Between Sponsors and the FDA. To improve collaboration between the Agency and sponsors, the commitment letter will likely include a new Biosimilar Biological Product Development (BPD) meeting type in which the Agency can provide focused targeted feedback. Modifications to the timelines and processes for Type 4 meetings are also likely.12
Regulatory Science Program. To facilitate more efficient biosimilar and interchangeable product development, as well as enhance regulatory decision-making, both the FDA and industry appeared to have agreed to funding a BsUFA Regulatory Science Program, modeled on the successful GDUFA Regulatory Science Program.13
Expedited Application Review. During negotiations, industry and the FDA discussed best practices for application review and opportunities for implementing those best practices into FDA documents and procedures.14
Next Steps
The BsUFA commitment letter is currently being reviewed by the Department of Health and Human Services (HHS), the Office of Personnel Management, and the Biden Administration. The BsUFA letter will also be open to public comment and discussed in a public meeting, likely to be held in mid-fall 2021, before being sent to Capitol Hill.
BsUFA III Negotiation Process
MDUFA
The MDUFA V negotiations have also been shaped by the experience of the FDA, industry, and other stakeholder groups during the COVID-19 pandemic. Both the FDA and industry want to keep elements of the regulatory flexibility that was provided during the pandemic as well as the increased amount of interaction between sponsors and the Agency. Additionally, for MDUFA V, both groups want to carry over many of the commitments and goals that were set during MDUFA IV. This approach aims to help maintain the status quo and ensure stability and continuity for the premarket review program in light of the many adjustments that the Agency had to make to manage the additional workload brought on by the COVID-19 pandemic. This approach will also provide additional time to achieve some of the commitments outlined in MDUFA IV that have not yet been met.15
Several proposals made by each group during the MDUFA V negotiations are still under discussion. These proposals include the following:
Hiring Targets and Vacancies. Throughout the negotiations there has been significant discussion regarding the number of vacancies for full-time equivalent (FTE) staff positions tied to user fees and the salaries for these positions. As part of MDUFA V, industry has proposed that annual specific numerical hiring targets be set in order to increase the formality of these goals, including increased transparency and prioritization by the Agency.16
Review of MDUFA IV One-Time Costs. Industry has proposed a review of funding for one-time costs from MDUFA IV including renewing funding for certain programs but not others. More specifically, industry would like to continue funding for “initiatives for patient engagement, recruitment, retention, and the independent assessment” while not renewing as part of MDUFA V funding “the investment to stand up time reporting” or “the IT investment to support digital health.” Other programs would require further discussion in order to be extended with MDUFA V funding, including “IT enhancements for premarket review work; real-world evidence; standards conformity assessment; and third party review.” In addition, industry has proposed to reinstate 5th year offset fees as the carryover balance has grown to a significant level.17
Device Safety. The FDA has presented a proposal to enhance its capabilities related to postmarket surveillance to allow the Agency “to more accurately and precisely identify the scope of potential concerns, to more efficiently resolve device performance and patient safety issues, and to provide timely and clear communications with patients and healthcare providers.”18 Although industry supports these enhanced capabilities, they noted during the negotiations that MDUFA funding has statutorily been limited to only premarket activities, and this significant change for industry-based funding would require statutory changes.
The Total Product Life Cycle Advisory Program (TAP). The FDA has presented the TAP program as a new model for frequent and rapid FDA interaction with sponsors that would also provide valuable feedback from external stakeholders such as payers and providers. The FDA described the program as including the hiring of new premarket review staff with different levels of expertise as well as inviting external stakeholder groups to participate in the process. The FDA also explained that the program aims to provide a more iterative engagement process with sponsors that could lead to higher quality submissions and fewer review cycles. Industry has voiced serious concern that many elements of the TAP program as currently outlined, such as convening private payers, seem to go beyond the scope of MDUFA and could require statutory changes. Industry is also concerned that this model could lead to more complex reviews and thereby increased program costs. Lastly, industry noted that there are already several FDA premarket programs that provide sponsors with increased engagement with the FDA as well as opportunities to discuss coverage with payers.19
Next Steps
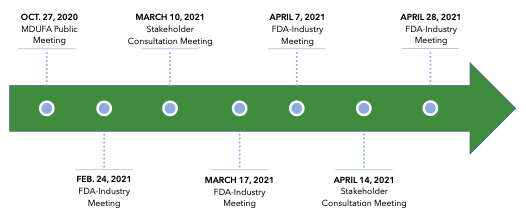
The FDA and industry will continue to meet throughout the coming year to develop an agreed-upon commitment letter to present to Congress. All stakeholder groups are encouraged to participate in the ongoing public negotiations. The most recent FDA-industry meeting was scheduled for May 19, 2021, but meeting minutes have not yet been published.20 Public records also show that several additional stakeholder consultation meetings were scheduled through the end of August.21
MDUFA V Negotiation Process22
1. Food and Drug Administration Website, FDA User Fee Programs, https://www.fda.gov/industry/fda-user-fee-programs.
2. Although not discussed in this memo, the Generic Drug User Fee Act (GDUFA) has the same end date and will likely be a part of the same legislative package as PDUFA, BsUFA, and MDUFA.
3. Derek Gingery, “PDUFA VII Negotiations Completed, Commitment Letter Ratification Ongoing,” Pink Sheet, 22 March 2021, https://pink.pharmaintelligence.informa.com/PS144034/PDUFA-VII-Negotiations-Completed-Commitment-Letter-Ratification-Ongoing.
4. FDA, PDUFA VII Commitment Letter, August 2021, https://www.fda.gov/media/151712/download.
5. PDUFA Reauthorization Meeting Summary, FDA and Industry Pre-Market Subgroup, 27 January 2021, https://www.fda.gov/media/147552/download.
6. PDUFA Reauthorization Meeting Summary, FDA and Industry CBER Breakout Subgroup, 10 November 2021, https://www.fda.gov/media/145588/download.
7. PDUFA Reauthorization Meeting Summary, FDA and Industry Pre-Market Subgroup, 21 January 2021, https://www.fda.gov/media/147564/download.
8. PDUFA Reauthorization Meeting Summary, FDA and Industry Negotiation Regulatory Decision Tools Subgroup, 1 December 2021, https://www.fda.gov/media/146389/download.
9. PDUFA Reauthorization Meeting Summary, FDA and Industry Digital Health and Informatics, 27 January 2021, https://www.fda.gov/media/146779/download.
10. PDUFA Reauthorization Meeting Summary, FDA and Industry Digital Health and Informatics, 16 December 2021, https://www.fda.gov/media/146775/download.
11. FDA, PDUFA VII Commitment Letter, August 2021, https://www.fda.gov/media/151712/download.
12. BSUFA Reauthorization Meeting Summary, FDA and Industry Steering Committee Meeting, 27 April 2021, https://www.fda.gov/media/149026/download.
13. BSUFA Reauthorization Meeting Summary, FDA and Industry Steering Committee Meeting, 13 April 2021, https://www.fda.gov/media/148185/download.
14. BSUFA Reauthorization Meeting Summary, FDA and Industry Steering Committee Meeting, 20 April 2021, https://www.fda.gov/media/149025/download.
15. Meeting Minutes, FDA–Industry MDUFA V Reauthorization Meeting, 28 April 2021, https://www.fda.gov/media/150625/download.
16. Ibid.
17. Ibid.
18. Ibid.
19. Ibid.
20. Ibid.
21. FDA Webpage, Stakeholder Consultation Meetings – Medical Device User Fee Amendments 2023 (MDUFA V), https://www.fda.gov/industry/medical-device-user-fee-amendments-mdufa/stakeholder-consultation-meetings-medical-device-user-fee-amendments-2023-mdufa-v.
22. FDA Webpage, Medical Device User Fee Amendments 2023 (MDUFA V), https://www.fda.gov/industry/medical-device-user-fee-amendments-mdufa/medical-device-user-fee-amendments-2023-mdufa-v.